<fieldset id="2sqqs"></fieldset>
<strike id="2sqqs"></strike>
文章來源:六月行研社
藥苑雜談?YAOYUANZATAN
藥物臨床試驗期間、藥品上市后都可以提出新增適應癥的申請,本文總結了幾種常見的新增適應癥的情形,從法規(guī)依據和注冊路徑兩方面概述,希望對大家有所啟發(fā)和幫助,但可能存在考慮不周全的地方,歡迎各位同行指正。
情形一:臨床試驗期間,提出新增適應癥
法規(guī)依據一:《藥品注冊管理辦法》
法規(guī)依據二:《化學藥品注冊受理審查指南》(第一部分 注冊分類1、2、5.1類)
對于新藥,同一藥物如有多個適應癥,按照一個適應癥對應一個IND/NDA提交注冊申請;雖然受理審查指南對于仿制藥(注冊分類3、4、5.2類的產品)未作相關說明,但如果仿制的參比制劑有多個適應癥,也應分別提交注冊申請。
注冊路徑:IND+NDA/ANDA。
藥品上市后新增適應癥主要分為三種情況:
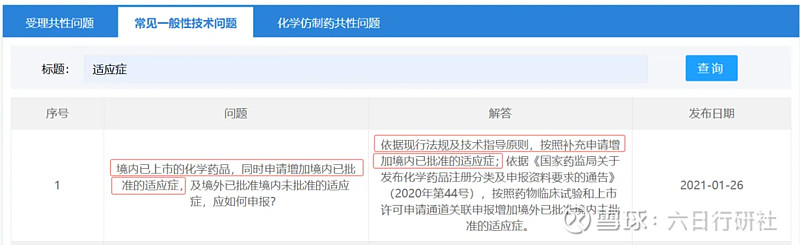
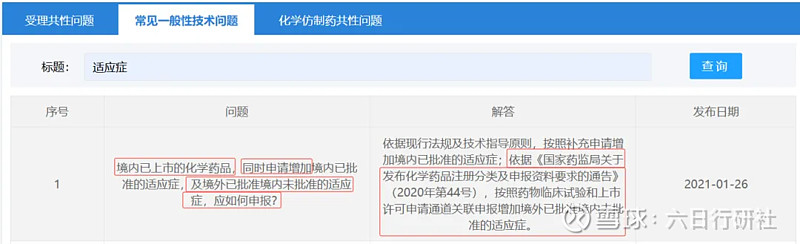
1)新增的適應癥為境內已批準的,即國內同品種已批準的適應癥(見情形二);
2)新增的適應癥為境外已批準但境內未批準的,此處已上市藥品包括仿制藥和新藥(分別見情形三、情形四);
3)新增的適應癥為境內和境外均未獲批的(情形五);
情形二:藥品上市后,新增的適應癥為境內
已批準的,即國內同品種已批準的適應癥
法規(guī)依據一:《已上市化學藥品和生物制品臨床變更技術指導原則》
法規(guī)依據二:CDE常見一般性技術問題
根據法規(guī)要求,情形二屬于重大變更,需要提交補充申請并經過審評、審批后執(zhí)行。因新增的適應癥為國內同品種已批準的,那么這個適應癥的安全性和有效性是明確的。對于普通口服固體制劑,如果能做到藥學一致、BE等效的話,大概率是不需要開展驗證性臨床。但如果是復雜制劑,比如復雜的注射劑,可能很難豁免,需具體問題具體分析。
注冊路徑:藥品上市許可持有人在開展臨床試驗前,應首先提出補充申請,在獲得批準后方可開展臨床試驗。藥品上市許可持有人完成臨床試驗并經評估認為試驗數(shù)據可支持相應變更時,可向國家藥品監(jiān)督管理局遞交補充申請。
情形三:藥品上市后(仿制藥),
新增的適應癥為境外已批準但境內未批準的
法規(guī)依據一:《化學藥品注冊受理審查指南》(第二部分 注冊分類3、4、5.2類)
注冊路徑:IND+ANDA(3類)
情形四:藥品上市后(新藥),
法規(guī)依據:《化學藥品注冊受理審查指南》(第一部分 注冊分類1、2、5.1類)
注冊路徑:IND+NDA(5.1類)
情形五:藥品上市后,
新增的適應癥為境內和境外均未獲批的
注冊路徑:IND+NDA(2.4類)
<fieldset id="ciyq2"></fieldset>